Arndt von Haeseler
Arndt von Haeseler's birthday is in 1959.
CIBIV-Website: www.cibiv.at
Campus Vienna Biocenter 5
1030 Vienna
Room: 1.107
![]() +43-1-4277-74307
+43-1-4277-74307
On this page
Q1: What are the evolutionary forces that have shaped the genomes of contemporary organisms and how can we infer the relevant parameters from multiple sequence alignments?
Q2: Can we develop mathematical, statistical and computational tools that help to analyse big data as generated by high throughput technologies in molecular biology?
Mathematical Models and efficient bioinformatics tools are the cornerstones to work on both questions. We develop such models and turn them into applicable software products for a wide user community. To understand the evolutionary forces, we are developing complex models of sequence evolution that in conjunction with tree reconstruction algorithms provide a comprehensive picture about the historical relationship of organisms and the changes that occur in a gene over time. Our approaches to Q2 are more diverse and are tailored to the special needs of high throughput technologies. We are interested in developing “stand-alone tools”, that can infer all relevant parameters from the input data.
Arndt von Haeseler's birthday is in 1959.
CIBIV-Website: www.cibiv.at
Campus Vienna Biocenter 5
1030 Vienna
Room: 1.107
![]() +43-1-4277-74307
+43-1-4277-74307
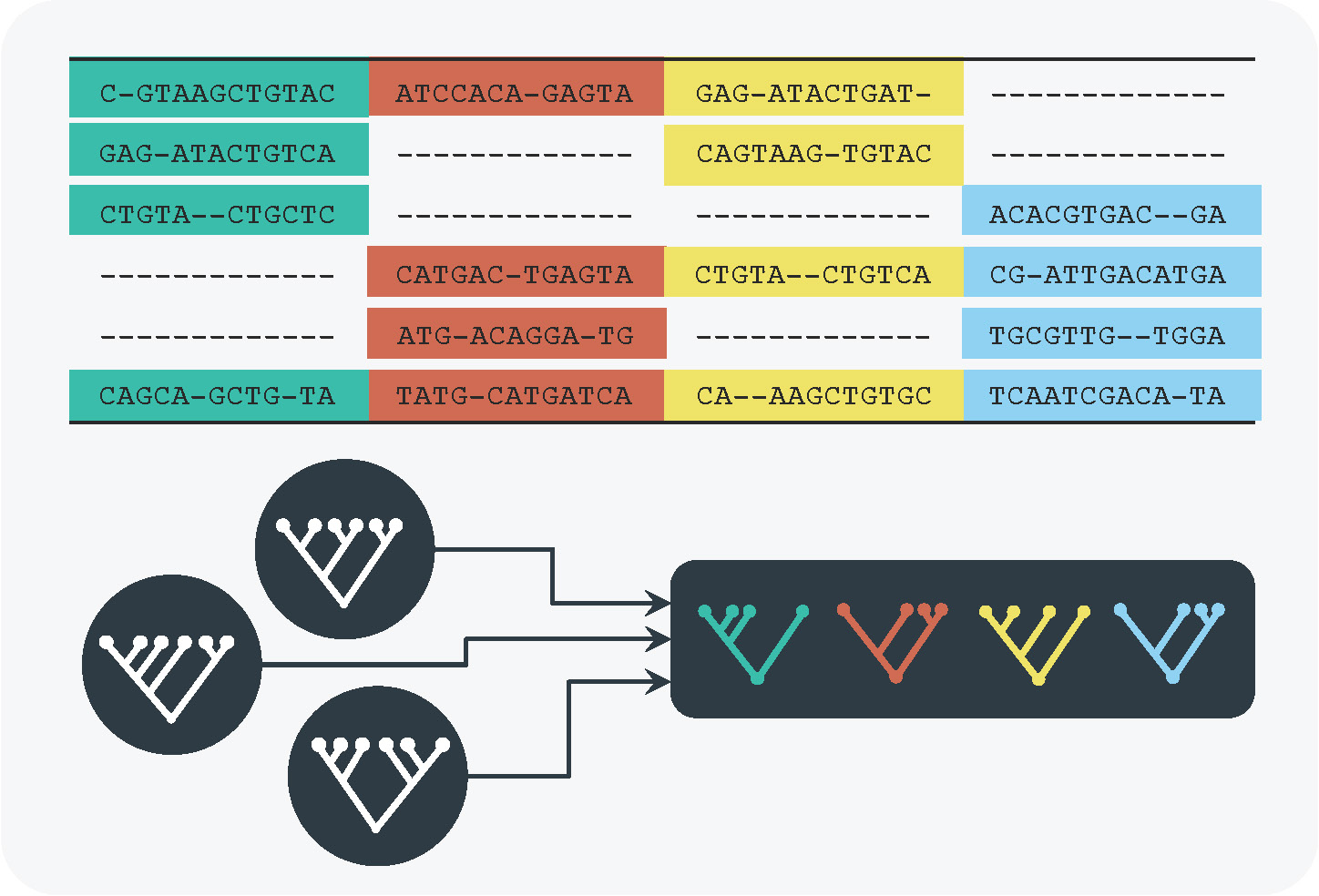
We developed an efficient tree reconstruction algorithm to take the patchy structure of genomic alignments into account and to infer large phylogenetic trees. These methods have been included in the widely used software tool IQ-TREE (http://www.iqtree.org).
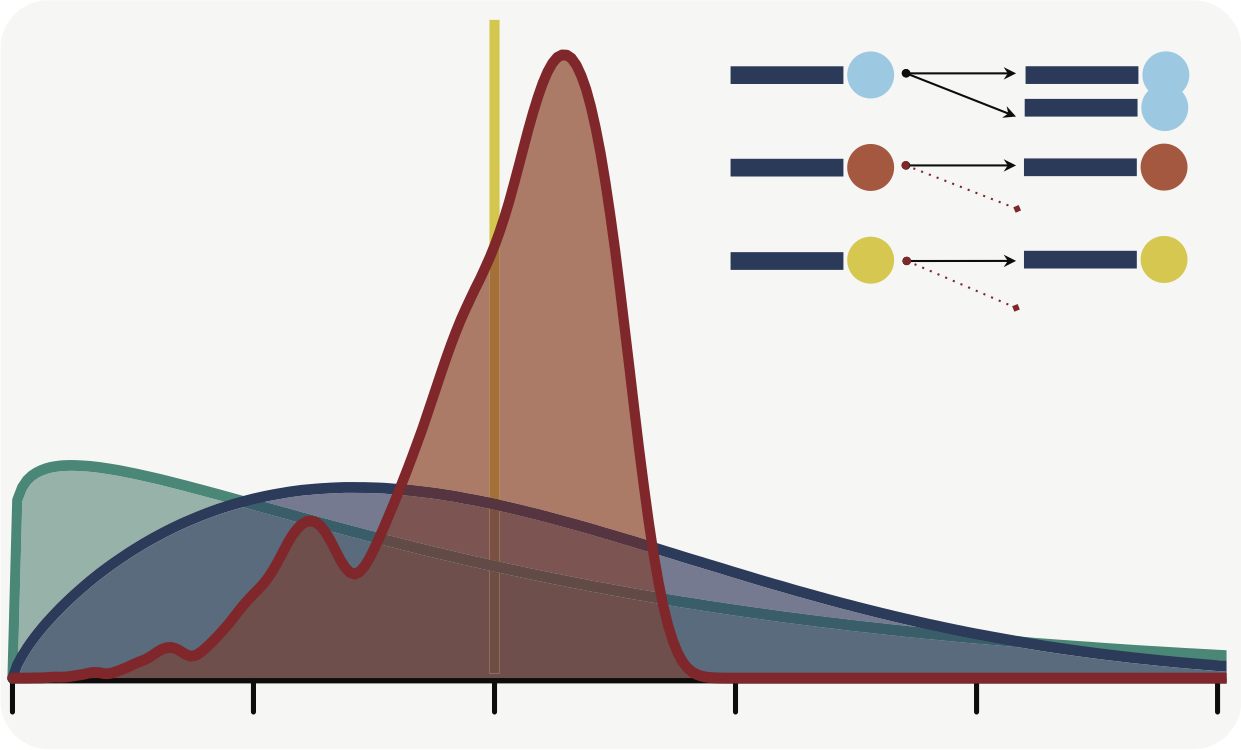
We tackled the following problem: For NGS experiments using unique molecular identifiers (UMIs), molecules that are lost entirely during sequencing cause under-estimation of the molecule count, and amplification artifacts like PCR chimeras cause over-estimation. TRUmiCount corrects UMI data for both types of errors, thus improving the accuracy of measured molecule counts considerably (https://cibiv.github.io/trumicount/).

Christiane Elgert
PostDoc ![]() +43-1-4277-74325
+43-1-4277-74325
Room: 1.812.4
Martin Fahrenberger
PhD Student ![]() +43-1-4277-74330
+43-1-4277-74330
Room: 1.812.2

Iris Gruber
Administration ![]() +43-1-4277-74320
+43-1-4277-74320
Room: 1.604
Simon Haendeler
PhD Student ![]() +43-1-4277-74329
+43-1-4277-74329
Room: 1.812.2
Robert Happel
IT Support ![]() +43-1-4277-74322
+43-1-4277-74322
Room: 1.812.1
Clemens Heiderer
Diploma/Master Student ![]() +43-1-4277-74320
+43-1-4277-74320
Room: 1.812.5
Laurenz Holcik
PhD Student ![]() +43-1-4277-74325
+43-1-4277-74325
Room: 1.812.3
Alina Leuchtenberger
PhD Student ![]() +43-1-4277-74330
+43-1-4277-74330
Room: 1.812.2
Julia Naas
PhD Student ![]() +43-1-4277-74328
+43-1-4277-74328
Room: 1.812.3

Cassius Manuel Perez de los Cobos Hermosa
PhD Student ![]() +43-1-4277-74326
+43-1-4277-74326
Room: 1.812.5

Enes Sakalli
PhD Student ![]() +43-1-4277-74320
+43-1-4277-74320
Room: 5.519

Heiko Schmidt
Senior PostDoc ![]() +43-1-4277-74321
+43-1-4277-74321
Room: 1.812.1

Arndt von Haeseler
Group Leader ![]() +43-1-4277-74307
+43-1-4277-74307
Room: 1.107
ModelFinder: fast model selection for accurate phylogenetic estimates.
Kalyaanamoorthy, Subha; Minh, Bui Quang; Wong, Thomas K F; von Haeseler, Arndt; Jermiin, Lars S
TRUmiCount: Correctly counting absolute numbers of molecules using unique molecular identifiers.
Pflug, Florian G; von Haeseler, Arndt
Next-generation sequencing diagnostics of bacteremia in septic patients.
Grumaz, Silke; Stevens, Philip; Grumaz, Christian; Decker, Sebastian O; Weigand, Markus A; Hofer, Stefan; Brenner, Thorsten; von Haeseler, Arndt; Sohn, Kai
NextGenMap: fast and accurate read mapping in highly polymorphic genomes.
Sedlazeck, Fritz J; Rescheneder, Philipp; von Haeseler, Arndt
The von Haeseler group participates in the special Doctoral Program "RNA Biology" reviewed and funded by the Austrian Research Fund FWF.
The Group Von Haeseler participates in the Special Research Area (SFB) "RNA-Reg - RNA regulation of the transcriptome" funded by the Austrian Science Fund FWF. SFB's are peer-reviewed, highly interactive research networks, established to foster long-term, interdisciplinary co-operation of local research groups working on the frontiers of their thematic areas.
The FWF project "Parallel computing for phylogenetic interference" funds an international collaborative effort to optimize and improve bioinformatic analysis methods for molecular data. The final aim of the project is to implement those new methods and models to be scalable on all modern multi-core, accelerator and supercomputer architectures.
The Group von Haeseler is an associated member of the special Doctoral Program "Signaling Mechanisms in Cellular Homeostasis" reviewed and funded by the Austrian Research Fund FWF.
AI for in vivo studies: Advantages of continuous monitoring of several animals in their home cage, without the use of RFID
 Sylvia Badurek - VBCF
Sylvia Badurek - VBCF Noah Weber
Noah WeberDate: 2024-07-11, Time: 10:00:00, Speaker: Noah Weber, Location: IMP Seminar Room 5.058, Type: Impromptu Seminar, Institute: Olden Labs, Host: Sylvia Badurek - VBCF
Monday Seminar (Internal)
None ZEILER Christina (Kovarik), BHARDWAJ Vishakha (Schlögelhofer)Date: 2024-07-15, Time: 12:30:00, Speaker: ZEILER Christina (Kovarik)BHARDWAJ Vishakha (Schlögelhofer), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
How the apoptotic factor Apaf1 behaves in cells during cell death
Elif Karagöz Wanda KukulskiDate: 2024-07-18, Time: 11:00:00, Speaker: Wanda Kukulski, Location: IMBA/GMI Lecture Hall, Type: VBC Regular Seminar, Institute: University of Bern, Host: Elif Karagöz
Monday Seminar (Internal)
None GUZMAN PEREZ Sebastian (Martinez), LITTLEBOY Jamie (Knoblich), ZMAJKOVIC Jakub (Zuber)Date: 2024-07-22, Time: 12:30:00, Speaker: GUZMAN PEREZ Sebastian (Martinez)LITTLEBOY Jamie (Knoblich)ZMAJKOVIC Jakub (Zuber), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Monday Seminar (Internal)
None MAURER Eric (Peters), DOCAVO GARCIA Antonio (Jachowicz), BABADEI Olga (Decker)Date: 2024-07-29, Time: 12:30:00, Speaker: MAURER Eric (Peters)DOCAVO GARCIA Antonio (Jachowicz)BABADEI Olga (Decker), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Monday Seminar (Internal)
None DEL CHIARO Alessia (Dagdas), HUETTER Christiane (Menche), SHETTY Kavya (Hein)Date: 2024-08-05, Time: 12:30:00, Speaker: DEL CHIARO Alessia (Dagdas)HUETTER Christiane (Menche)SHETTY Kavya (Hein), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
An alternative mechanism to form an organizing center in mouse neural tube organoids
None Teresa KrammerDate: 2024-08-08, Time: 15:00:00, Speaker: Teresa Krammer, Location: IMBA/GMI Lecture Hall, Type: PhD Defense, Institute: IMBA - Tanaka Lab, Host: None
Advanced Concepts of Super-Resolution Fluorescence Microscopy
Francisco Balzarotti Jörg EnderleinDate: 2024-08-09, Time: 10:00:00, Speaker: Jörg Enderlein, Location: IMP Lecture Hall, Type: IMP Impromptu Seminar, Institute: Georg August University, Host: Francisco Balzarotti
Monday Seminar (Internal)
None ANTONIOLI Sumire (Leonard), EMTENANI Shamsi(Knoblich), SANDRU Ana Maria (Baccarini)Date: 2024-08-12, Time: 12:30:00, Speaker: ANTONIOLI Sumire (Leonard)EMTENANI Shamsi(Knoblich)SANDRU Ana Maria (Baccarini), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Monday Seminar (Internal)
BENEDUM Johannes (Slade), KALIS Robert (Zuber), PATEL Rajvi (Matos)Date: 2024-08-19, Time: 12:30:00, Speaker: BENEDUM Johannes (Slade)KALIS Robert (Zuber)PATEL Rajvi (Matos), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host:
Monday Seminar (Internal)
None JANSEN Ralf (Brennecke), SILVA Raquel (Foisner), BASSAT Elad (Tanaka)Date: 2024-08-26, Time: 12:30:00, Speaker: JANSEN Ralf (Brennecke)SILVA Raquel (Foisner)BASSAT Elad (Tanaka), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Monday Seminar (Internal)
None SCHAAR Benjamin (Zimmer), IGOLKINA Anna (Nordborg), PROTSENKO Liudmila (Brennecke)Date: 2024-09-02, Time: 12:30:00, Speaker: SCHAAR Benjamin (Zimmer)IGOLKINA Anna (Nordborg)PROTSENKO Liudmila (Brennecke), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Striking physiology and cell biology of (marine) environmental microorganisms
Kristin Tessmar-Raible Ralf RabusDate: 2024-09-03, Time: 11:30:00, Speaker: Ralf Rabus, Location: Max Perutz Labs SR 1 (6th floor), Type: New Developments in Chromosome Biology, Institute: ICBM, Carl von Ossietzky Universität Oldenburg, Host: Kristin Tessmar-Raible
Title to be announced
Daniel Gerlich Alexander Van OudenaardenDate: 2024-09-05, Time: 11:00:00, Speaker: Alexander Van Oudenaarden, Location: IMBA/GMI Lecture Hall, Type: VBC Regular Seminar, Institute: Hubrecht Institute, Host: Daniel Gerlich
Monday Seminar (Internal)
STUART Hannah (Tanaka), ROVERA Emanuele (Zuber), KLOSS Linda (Technau)Date: 2024-09-09, Time: 12:30:00, Speaker: STUART Hannah (Tanaka)ROVERA Emanuele (Zuber)KLOSS Linda (Technau), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host:
Mechanisms controlling maintenance of cohesin dependent loops
Franz Klein Frederic BeckouetDate: 2024-09-10, Time: 11:30:00, Speaker: Frederic Beckouet, Location: Max Perutz Labs SR 1 (6.501), Type: SFB Seminar, Institute: Toulouse Centre de Biologie Intégrative, Host: Franz Klein
Title to be announced
Silvia Ramundo, Fred Berger Elcin ÜnalDate: 2024-09-12, Time: 11:00:00, Speaker: Elcin Ünal, Location: IMBA/GMI Lecture Hall, Type: VBC Regular Seminar, Institute: UC Berkeley, Host: Silvia Ramundo, Fred Berger
Monday Seminar (Internal)
WUN Cheuk Ling (Ramundo), MÜLLER Marlene (Rivron), RAUSCHMEIER Rene (Van der Veeken)Date: 2024-09-16, Time: 12:30:00, Speaker: WUN Cheuk Ling (Ramundo)MÜLLER Marlene (Rivron)RAUSCHMEIER Rene (Van der Veeken), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host:
Title to be announced
Marco Hein Noam Stern-GinossarDate: 2024-09-19, Time: 11:00:00, Speaker: Noam Stern-Ginossar, Location: IMBA/GMI Lecture Hall, Type: VBC Regular Seminar, Institute: Weizmann Institute of Science, Host: Marco Hein
Monday Seminar (Internal)
ESTIVILL I CAPARROS Guillem (Obenauf), VOJTEK Matus (Stark), DA COSTA Barbara (Berger)Date: 2024-09-23, Time: 12:30:00, Speaker: ESTIVILL I CAPARROS Guillem (Obenauf)VOJTEK Matus (Stark)DA COSTA Barbara (Berger), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host:
Title to be announced
Joao Matos Maria MotaDate: 2024-09-26, Time: 11:00:00, Speaker: Maria Mota, Location: IMBA/GMI Lecture Hall, Type: VBC Regular Seminar, Institute: University of Lisbon, Host: Joao Matos
Title to be announced
Julius Brennecke Deborah Bourc'hisDate: 2024-09-27, Time: 11:00:00, Speaker: Deborah Bourc'his, Location: IMBA/GMI Lecture Hall, Type: Impromptu Seminar, Institute: Institut Curie, Host: Julius Brennecke
Monday Seminar (Internal)
JÄGER Christina (Van der Veeken), SHUKLA Vikas (Berger), RODRIGUEZ TERRONES Diego (Tanaka)Date: 2024-09-30, Time: 12:30:00, Speaker: JÄGER Christina (Van der Veeken)SHUKLA Vikas (Berger)RODRIGUEZ TERRONES Diego (Tanaka), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host:
Monday Seminar (Internal)
KANNO Tatsuo (Berger), SCHIMMER Clara (Ellis), DE SANTIS Martina (Rivron)Date: 2024-10-07, Time: 12:30:00, Speaker: KANNO Tatsuo (Berger)SCHIMMER Clara (Ellis)DE SANTIS Martina (Rivron), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host:
Title to be announced
Stephanie Ellis Cedric BlanpainDate: 2024-10-10, Time: 11:00:00, Speaker: Cedric Blanpain, Location: , Type: , Institute: Universite Libres Bruxelles, Host: Stephanie Ellis
Monday Seminar (Internal)
None GAO Peng (Dagdas), HANDLER Dominik (Brennecke), FRIDRICH Arie (Berger)Date: 2024-10-14, Time: 12:30:00, Speaker: GAO Peng (Dagdas)HANDLER Dominik (Brennecke)FRIDRICH Arie (Berger), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Title to be announced
Alex Stark, Francisco Balzarotti Ralf JungmannDate: 2024-10-17, Time: 11:00:00, Speaker: Ralf Jungmann, Location: IMBA/GMI Lecture Hall, Type: VBC Regular Seminar, Institute: LMU, MPI, Host: Alex Stark, Francisco Balzarotti
Monday Seminar (Internal)
MILOJKOVIC Lidija (Urban), SPENCER Victoria (Dolan), AUGUSTO Pedro (Zimmer)Date: 2024-10-21, Time: 12:30:00, Speaker: MILOJKOVIC Lidija (Urban)SPENCER Victoria (Dolan)AUGUSTO Pedro (Zimmer), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host:
Title to be announced
Moritz Gaidt Andrea AblasserDate: 2024-10-24, Time: 11:00:00, Speaker: Andrea Ablasser, Location: IMP Lecture Hall, Type: VBC Regular Seminar, Institute: EPFL, Host: Moritz Gaidt
Monday Seminar (Internal)
None GOMEZ SEGALAS Alba (Balzarotti), KLEINFERCHER Sebastian (Stark), PRADHAN Saurabh (Rivron)Date: 2024-10-28, Time: 12:30:00, Speaker: GOMEZ SEGALAS Alba (Balzarotti)KLEINFERCHER Sebastian (Stark)PRADHAN Saurabh (Rivron), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Monday Seminar (Internal)
SCHEPERS Sandra (Knoblich), BUSETTO Virginia (Falk), DMITRIEV Vladimir (Goloborodko)Date: 2024-11-04, Time: 12:30:00, Speaker: SCHEPERS Sandra (Knoblich)BUSETTO Virginia (Falk)DMITRIEV Vladimir (Goloborodko), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host:
VBC PhD Symposium
NoneDate: 2024-11-07, Time: 09:00:00, Speaker: , Location: IMP Lecture Hall, Type: Symposium, Institute: None, Host: None
Monday Seminar (Internal)
GARCIA GALLARDO Maria (Bücker), HARVEY Zachary (Berger), SAKALLI Enes (Von Haeseler,Menche)Date: 2024-11-11, Time: 12:30:00, Speaker: GARCIA GALLARDO Maria (Bücker)HARVEY Zachary (Berger)SAKALLI Enes (Von Haeseler,Menche), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host:
Title to be announced
Sasha Menjdan Benoit BruneauDate: 2024-11-14, Time: 11:00:00, Speaker: Benoit Bruneau, Location: IMBA/GMI Lecture Hall, Type: VBC Regular Seminar, Institute: Gladstone Institute of Cardiovascular Disease, UCSF, Host: Sasha Menjdan
Title to be announced
Alex Stark Lacramioara BintuDate: 2024-11-18, Time: 11:00:00, Speaker: Lacramioara Bintu, Location: IMBA/GMI Lecture Hall, Type: Impromptu Seminar, Institute: Stanford University, Host: Alex Stark
Monday Seminar (Internal)
None FESSELET Jeanne (Kovarik), PAPAREDDY Ranjith (Dagdas), SHEN Jingyi (Obenauf)Date: 2024-11-18, Time: 12:30:00, Speaker: FESSELET Jeanne (Kovarik)PAPAREDDY Ranjith (Dagdas)SHEN Jingyi (Obenauf), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Title to be announced
Irma Querques Martin JinekDate: 2024-11-21, Time: 11:00:00, Speaker: Martin Jinek, Location: IMBA/GMI Lecture Hall, Type: VBC Regular Seminar, Institute: University of Zurich, Host: Irma Querques
Monday Seminar (Internal)
BOREIKAITE Vytaute (Plaschka), SRINIVASAN Navaneeth (Peters), DODOMANI Ananya (Campbell)Date: 2024-11-25, Time: 12:30:00, Speaker: BOREIKAITE Vytaute (Plaschka)SRINIVASAN Navaneeth (Peters)DODOMANI Ananya (Campbell), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host:
Title to be announced
Alwin Köhler David BakerDate: 2024-11-28, Time: 18:00:00, Speaker: David Baker, Location: Zoom, Type: VBC Regular Seminar via Zoom, Institute: UW Dept. of Biochemistry, Host: Alwin Köhler
Monday Seminar (Internal)
None SOMMER Theresa (Rivron), , MAIR Zahar (Dolan)Date: 2024-12-02, Time: 12:30:00, Speaker: SOMMER Theresa (Rivron)MAIR Zahar (Dolan), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Title to be announced
Joao Matos Ian HicksonDate: 2024-12-05, Time: 11:00:00, Speaker: Ian Hickson, Location: IMBA/GMI Lecture Hall, Type: VBC Regular Seminar, Institute: University of Copenhagen, Host: Joao Matos
Monday Seminar (Internal)
None BREHM Martin (Falk), PLATZER Sebastian (Hein), RAFANEL Baptiste (Brennecke)Date: 2024-12-09, Time: 12:30:00, Speaker: BREHM Martin (Falk)PLATZER Sebastian (Hein)RAFANEL Baptiste (Brennecke), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Title to be announced
Joao Matos Stephen WestDate: 2024-12-12, Time: 11:00:00, Speaker: Stephen West, Location: IMBA/GMI Lecture Hall, Type: VBC Regular Seminar, Institute: The Francis Crick Institute, Host: Joao Matos
Monday Seminar (Internal)
None DAHAN Tal (Nordborg), CHUGUNOVA Anastasia (Pauli), VON WIREN Julius (Kovarik)Date: 2024-12-16, Time: 12:30:00, Speaker: DAHAN Tal (Nordborg)CHUGUNOVA Anastasia (Pauli)VON WIREN Julius (Kovarik), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Title to be announced
Anna Obenauf Uri Ben-DavidDate: 2025-01-09, Time: 11:00:00, Speaker: Uri Ben-David, Location: IMBA/GMI Lecture Hall, Type: VBC Regular Seminar, Institute: Tel Aviv University, Host: Anna Obenauf
Monday Seminar (Internal)
KRASNOVID Filipp (Mari-Ordonez), ROTHE Patricia (Plaschka), KROGULL Daniel (Burga)Date: 2025-01-13, Time: 12:30:00, Speaker: KRASNOVID Filipp (Mari-Ordonez)ROTHE Patricia (Plaschka)KROGULL Daniel (Burga), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host:
Monday Seminar (Internal)
VICIC Ziga (Jachowicz), GRÜNEWALD Jakob (Obenauf), MANOLOVA Toni (Falk)Date: 2025-01-20, Time: 12:30:00, Speaker: VICIC Ziga (Jachowicz)GRÜNEWALD Jakob (Obenauf)MANOLOVA Toni (Falk), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host:
Title to be announced
Egon Ogris Anne BertolottiDate: 2025-01-23, Time: 11:00:00, Speaker: Anne Bertolotti, Location: IMBA/GMI Lecture Hall, Type: VBC Regular Seminar, Institute: MCR Laboratory of Molecular Biology, Host: Egon Ogris
Monday Seminar (Internal)
SCARAMUZZA Federico (Tessmar), WALTER Mateusz (Querques), WILLIAMS Thomas (Clausen)Date: 2025-01-27, Time: 12:30:00, Speaker: SCARAMUZZA Federico (Tessmar) WALTER Mateusz (Querques)WILLIAMS Thomas (Clausen), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host:
Monday Seminar (Internal)
CRUZ Joseph (Gerlich), GHEORGHITA Andreea (Clausen), GALUSKA Philipp (Djinovic)Date: 2025-02-03, Time: 12:30:00, Speaker: CRUZ Joseph (Gerlich)GHEORGHITA Andreea (Clausen)GALUSKA Philipp (Djinovic), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host:
Monday Seminar (Internal)
None GRIGOREVA Elizaveta (Nordborg), KOHOUTKOVA Eliska (Clausen), STEINACKER Thomas (Gerlich)Date: 2025-02-10, Time: 12:30:00, Speaker: GRIGOREVA Elizaveta (Nordborg)KOHOUTKOVA Eliska (Clausen)STEINACKER Thomas (Gerlich), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Title to be announced
Fred Berger Jeff BoekeDate: 2025-02-13, Time: 11:00:00, Speaker: Jeff Boeke, Location: IMBA/GMI Lecture Hall, Type: VBC Regular Seminar, Institute: NYU Langdone Health, Host: Fred Berger
Monday Seminar (Internal)
None RAFFL Gerald (Ameres), VULIN Milica (Obenauf), FISCHER Andre (Tanaka)Date: 2025-02-17, Time: 12:30:00, Speaker: RAFFL Gerald (Ameres)VULIN Milica (Obenauf)FISCHER Andre (Tanaka), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Monday Seminar (Internal)
None BAYERL Jonas (Zuber), ZABOLOCKI Michael (Knoblich), EDER Stephanie (Zimmer)Date: 2025-02-24, Time: 12:30:00, Speaker: BAYERL Jonas (Zuber)ZABOLOCKI Michael (Knoblich)EDER Stephanie (Zimmer), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Title to be announced
Joanna Jachowicz Cedric FeschotteDate: 2025-02-27, Time: 11:00:00, Speaker: Cedric Feschotte, Location: IMBA/GMI Lecture Hall, Type: VBC Regular Seminar, Institute: Cornell University, Host: Joanna Jachowicz
Monday Seminar (Internal)
None KROETENHEERDT Eva (Leonard), RÖHSNER Josef (Pauli), RIABOV Daria (Plaschka)Date: 2025-03-03, Time: 12:30:00, Speaker: KROETENHEERDT Eva (Leonard)RÖHSNER Josef (Pauli)RIABOV Daria (Plaschka), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Monday Seminar (Internal)
None CHO Chung Hyun (Berger), SEIDL Carina (Tanaka), MIGOTTI Mario (Konrat)Date: 2025-03-10, Time: 12:30:00, Speaker: CHO Chung Hyun (Berger)SEIDL Carina (Tanaka)MIGOTTI Mario (Konrat), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Monday Seminar (Internal)
None CHEN Shenzhi (Stark), BINDL Michael (Leeb), JÄGER Martin (Obenauf)Date: 2025-03-17, Time: 12:30:00, Speaker: CHEN Shenzhi (Stark)BINDL Michael (Leeb)JÄGER Martin (Obenauf), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Title to be announced
Joao Matos Geert KopsDate: 2025-03-20, Time: 11:00:00, Speaker: Geert Kops, Location: IMBA/GMI Lecture Hall, Type: VBC Regular Seminar, Institute: Hubrecht Institute, Host: Joao Matos
Monday Seminar (Internal)
None MAYA Juan (Balzarotti), DOSA Anna (Clausen), KOBER Julia (Ramundo)Date: 2025-03-24, Time: 12:30:00, Speaker: MAYA Juan (Balzarotti)DOSA Anna (Clausen)KOBER Julia (Ramundo), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Title to be announced
Alwin Köhler Martin BeckDate: 2025-03-27, Time: 11:00:00, Speaker: Martin Beck, Location: IMBA/GMI Lecture Hall, Type: VBC Regular Seminar, Institute: Max Planck Institute for Biophysics, Host: Alwin Köhler
Monday Seminar (Internal)
None HOLZER Elisabeth (Martens), THEMANN Jan (Knoblich), GROSS Angelina (Dagdas)Date: 2025-03-31, Time: 12:30:00, Speaker: HOLZER Elisabeth (Martens)THEMANN Jan (Knoblich)GROSS Angelina (Dagdas), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Monday Seminar (Internal)
None GROH Roan (Dagdas), CHAO Victor (Huis)Date: 2025-04-07, Time: 12:30:00, Speaker: GROH Roan (Dagdas)CHAO Victor (Huis), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Monday Seminar (Internal)
None OKUN Anastasia (Karagöz), COLLISON Robert (Ramundo), WELZL Paul (Balzarotti/Plaschka)Date: 2025-04-14, Time: 12:30:00, Speaker: OKUN Anastasia (Karagöz)COLLISON Robert (Ramundo)WELZL Paul (Balzarotti/Plaschka), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Title to be announced
Anton Goloborodko Maria Cristina GambettaDate: 2025-04-24, Time: 11:00:00, Speaker: Maria Cristina Gambetta, Location: IMBA/GMI Lecture Hall, Type: VBC Regular Seminar, Institute: University of Lausanne, Host: Anton Goloborodko
Monday Seminar (Internal)
None SUWITA Johannes (Pauli), GRABARCZYK Daniel (Clausen), PANDA Aswini (Falk)Date: 2025-04-28, Time: 12:30:00, Speaker: SUWITA Johannes (Pauli)GRABARCZYK Daniel (Clausen)PANDA Aswini (Falk), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Monday Seminar (Internal)
None KIM Olena (Knoblich), JOB Nikhil (Dolan), ADELMANN Leonie (Raible)Date: 2025-05-05, Time: 12:30:00, Speaker: KIM Olena (Knoblich)JOB Nikhil (Dolan)ADELMANN Leonie (Raible), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Microsymposium on Small RNA Biology
NoneDate: 2025-05-07, Time: 09:00:00, Speaker: , Location: IMP Lecture Hall, Type: Symposium, Institute: None, Host: None
Monday Seminar (Internal)
None SCIBISZ Grzegorz (Karagöz), VAN GENDEREN Emiel (Rivron), BOZKURT Miray (Tanaka)Date: 2025-05-12, Time: 12:30:00, Speaker: SCIBISZ Grzegorz (Karagöz)VAN GENDEREN Emiel (Rivron)BOZKURT Miray (Tanaka), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Title to be announced
Alex Stark Chuna Ram ChoudharyDate: 2025-05-15, Time: 11:00:00, Speaker: Chuna Ram Choudhary, Location: IMBA/GMI Lecture Hall, Type: VBC Regular Seminar, Institute: University of Copenhagen, Host: Alex Stark
Monday Seminar (Internal)
None POTTENDORFER Elisabeth (Obenauf), WALLNER Eva-Sophie (Dolan), BECKER Moritz (Leeb)Date: 2025-05-19, Time: 12:30:00, Speaker: POTTENDORFER Elisabeth (Obenauf)WALLNER Eva-Sophie (Dolan)BECKER Moritz (Leeb), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Title to be announced
Elif Karagöz Manuel LeonettiDate: 2025-05-22, Time: 11:00:00, Speaker: Manuel Leonetti, Location: IMBA/GMI Lecture Hall, Type: VBC Regular Seminar, Institute: CZ Biohub San Francisco, Host: Elif Karagöz
Monday Seminar (Internal)
None DIMITROVA Polina (Van der Veeken), STRAVS Ana (Knoblich), SCHÖNLEIN Martin (Obenauf)Date: 2025-05-26, Time: 12:30:00, Speaker: DIMITROVA Polina (Van der Veeken)STRAVS Ana (Knoblich)SCHÖNLEIN Martin (Obenauf), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Monday Seminar (Internal)
None SABATH Kevin (Plaschka/Stark), MANCHENO JUNCOSA Estela (Mendjan), HORENKAMP Simone (Tanaka)Date: 2025-06-02, Time: 12:30:00, Speaker: SABATH Kevin (Plaschka/Stark)MANCHENO JUNCOSA Estela (Mendjan)HORENKAMP Simone (Tanaka), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Monday Seminar (Internal)
None BERGER Sonja (Goloborodko), KOWALD Saskia (Gaidt), KAPRAL Thomas (Zagrovic)Date: 2025-06-16, Time: 12:30:00, Speaker: BERGER Sonja (Goloborodko)KOWALD Saskia (Gaidt)KAPRAL Thomas (Zagrovic), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Monday Seminar (Internal)
None BUCHERON Chloe (Menche), KRUMWIEDE Luisa (Gaidt), PACHANO Tomas (Stark)Date: 2025-06-23, Time: 12:30:00, Speaker: BUCHERON Chloe (Menche)KRUMWIEDE Luisa (Gaidt)PACHANO Tomas (Stark), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Title to be announced
Joris Van der Veeken Nikhil JoshiDate: 2025-06-26, Time: 11:00:00, Speaker: Nikhil Joshi, Location: IMBA/GMI Lecture Hall, Type: VBC Regular Seminar, Institute: Yale University, Host: Joris Van der Veeken
Monday Seminar (Internal)
None SCHINDLER Lukas (Schlögelhofer), MANDLBURGER Nikolaus (Stark), OTTO Magdalena (Leonard)Date: 2025-06-30, Time: 12:30:00, Speaker: SCHINDLER Lukas (Schlögelhofer)MANDLBURGER Nikolaus (Stark)OTTO Magdalena (Leonard), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Title to be announced
Marco Hein Matthias MannDate: 2025-07-03, Time: 11:00:00, Speaker: Matthias Mann, Location: IMBA/GMI Lecture Hall, Type: VBC Regular Seminar, Institute: Max-Planck Institute of Biochemistry, Host: Marco Hein
Monday Seminar (Internal)
None TRUS Palina (Peters), KOBAKHIDZE George (Dong), KONGSTED Thea (Dolan)Date: 2025-07-07, Time: 12:30:00, Speaker: TRUS Palina (Peters)KOBAKHIDZE George (Dong)KONGSTED Thea (Dolan), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Title to be announced
Erinc Hallacli Simon AlbertiDate: 2025-07-10, Time: 11:00:00, Speaker: Simon Alberti, Location: IMBA/GMI Lecture Hall, Type: VBC Regular Seminar, Institute: TU Dresden, Host: Erinc Hallacli
Monday Seminar (Internal)
None RUSCH Emma (Golobordko), VELIKOV Daniel (Matos), VOGEL Alexander (Clausen/Yudushkin)Date: 2025-07-14, Time: 12:30:00, Speaker: RUSCH Emma (Golobordko)VELIKOV Daniel (Matos)VOGEL Alexander (Clausen/Yudushkin), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Monday Seminar (Internal)
None ANGELOPOULOU Eleni (Ellis), SCHAFFER Petra (Grade), RAMIREZ America (Köhler)Date: 2025-07-21, Time: 12:30:00, Speaker: ANGELOPOULOU Eleni (Ellis)SCHAFFER Petra (Grade)RAMIREZ America (Köhler), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Monday Seminar (Internal)
None DIMITRIADI Anna (Mendjan), LEITNER Moritz (Martinez), RAUH Felix (Stark)Date: 2025-07-28, Time: 12:30:00, Speaker: DIMITRIADI Anna (Mendjan)LEITNER Moritz (Martinez)RAUH Felix (Stark), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Monday Seminar (Internal)
None LI Wenxin (Zimmer), FISCHER Felix (Zagrovic), ABESAMIS Kim Ivan (Dong)Date: 2025-08-04, Time: 12:30:00, Speaker: LI Wenxin (Zimmer)FISCHER Felix (Zagrovic)ABESAMIS Kim Ivan (Dong), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Monday Seminar (Internal)
None KLEIFELD Justus (Urban), MCATEER Tara (Pinheiro), BHATTACHARYA Mrinnanda (Zimmer)Date: 2025-08-11, Time: 12:30:00, Speaker: KLEIFELD Justus (Urban)MCATEER Tara (Pinheiro)BHATTACHARYA Mrinnanda (Zimmer), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Monday Seminar (Internal)
None BELL Lillie (Clausen), MIHAILOVIC Milica (Karagöz), GUYNES Keroshini (Jachowicz)Date: 2025-08-18, Time: 12:30:00, Speaker: BELL Lillie (Clausen)MIHAILOVIC Milica (Karagöz)GUYNES Keroshini (Jachowicz), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Monday Seminar (Internal)
None BOHL VIALLEFONT Gregoire (Nordborg), MARCHIORI Elisa (Van der Veeken), JAHNEL Stefan (Mendjan)Date: 2025-08-25, Time: 12:30:00, Speaker: BOHL VIALLEFONT Gregoire (Nordborg)MARCHIORI Elisa (Van der Veeken)JAHNEL Stefan (Mendjan), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Monday Seminar (Internal)
None KIRCHGATTERER Paul (Gaidt), ACHLEITNER Sonja (Martens), KAYA Oguzhan (Knoblich)Date: 2025-09-01, Time: 12:30:00, Speaker: KIRCHGATTERER Paul (Gaidt)ACHLEITNER Sonja (Martens)KAYA Oguzhan (Knoblich), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Monday Seminar (Internal)
None HUTH Michelle (Leeb), RADHAKRISHNA PILLAI Balashankar (Brennecke), REN Kaike (Otsuka)Date: 2025-09-08, Time: 12:30:00, Speaker: HUTH Michelle (Leeb)RADHAKRISHNA PILLAI Balashankar (Brennecke)REN Kaike (Otsuka), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Monday Seminar (Internal)
None GONZALEZ ALVARADO Maria Nazareth (Grade), KASHKO Nataliia (Matos), OBERHUEMER Michael (Leeb)Date: 2025-09-15, Time: 12:30:00, Speaker: GONZALEZ ALVARADO Maria Nazareth (Grade)KASHKO Nataliia (Matos)OBERHUEMER Michael (Leeb), Location: IMP Lecture Hall, Type: Monday Seminar, Institute: Open only to IMP/IMBA/GMI/Max Perutz Labs, Host: None
Title to be announced
Joao Matos, Peter Schlögelhofer Katja WassmannDate: 2025-09-18, Time: 11:00:00, Speaker: Katja Wassmann, Location: IMBA/GMI Lecture Hall, Type: VBC Regular Seminar, Institute: Institute Jacques Monod, Host: Joao Matos, Peter Schlögelhofer
Title to be announced
Andrea Pauli Anne BrunetDate: 2025-10-16, Time: 11:00:00, Speaker: Anne Brunet, Location: IMBA/GMI Lecture Hall, Type: VBC Regular Seminar, Institute: Stanford University, Host: Andrea Pauli
Title to be announced
David Haselbach Kresten Lindorff-LarssenDate: 2025-10-30, Time: 11:00:00, Speaker: Kresten Lindorff-Larssen, Location: IMBA/GMI Lecture Hall, Type: VBC Regular Seminar, Institute: University of Copenhagen, Host: David Haselbach